流行病学
流行病学
流行病学:早在18世纪Schloein就提出了血友病这一概念。1893年Wright首次发现血友病患者的凝血时间延长,认为本病是原发性出血性疾病。1947年人们认识到血友病患者的凝血时间延长与FⅧ水平下降有关,随后于1952年发现
Christmas病是由于FIX水平下降所致,之后将FⅧ水平下降的血友病命名为
血友病甲,将
Christmas病命名为
血友病乙。
在20世纪30年代发现正常的血浆能够纠正血友病患者的凝血时间延长,随后正常血浆替代治疗成为早期治疗血友病的基本方法。但是血浆中因子含量往往不能满足治疗的需要,因出血导致的死亡率仍很高。1964年冷沉淀物的发现开创了血友病治疗的新时代,冷沉淀物中含有丰富的FⅧ,可用于
血友病甲患者的治疗。1965年出现了FⅧ部分纯化制剂,称为FⅧ浓缩剂(主要含有FⅧ和vWF)。自20世纪80年代始,为了防止输血相关的病毒感染,从献血者的筛选、因子浓缩制剂的病毒灭活处理以及纯化工艺等多方面,对替代治疗的产品进行了改进。1985年FⅧ和FⅨ基因克隆成功,1989年基因重组的FⅧ首次投入临床使用,近几年高纯度FⅨ制剂和基因重组的FⅨ也已开始使用。
血友病的发病率约为1/5000男性人口,无明显地区和种族差异。其中
血友病甲约占80%~85%,
血友病乙约占10%~15%。据1992年24省、直辖市调查血友病的患病率为2.73/10万。
血友病甲、乙、丙的发病比率约为16∶3∶1。其共同特点为终身有
自发的或轻微损伤后长时间出血倾向。
发病机制
发病机制:FⅧ和FⅨ基因均定位于X染色体长臂末端,因此
血友病甲和
血友病乙都是X染色体连锁的遗传性疾病。根据血友病的遗传规律可以有以下4种情况:
1.男性患者与正常女性所生的男孩均是正常者,所生的女孩均是携带者。
2.女性携带者与正常男性所生的男孩有50%的概率为血友病患者,所生的女孩有50%的概率是携带者。
3.女性携带者和男性患者所生的男孩有50%的概率是血友病患者,所生女孩携带者和血友病患者各占50%,此种婚配情况很少见。
4.男性血友病患者和女性血友病患者所生的男孩和女孩均患血友病,此种婚配情况至今尚未发现一例。
因子Ⅷ是一种大分子复合物,在血浆中由小分子量具有促凝血活性的Ⅷ:C和大分子量的von Willebrand因子(vWF)以非共价键的形式相结合形成复合物,其中Ⅷ∶C只占复合物的1%。因子Ⅷ是一种水溶性糖蛋白,可被Xa或凝血酶激活为Ⅷa。内源性凝血系统中,在Ca2 及磷脂存在的条件下,Ⅷa以辅酶的形式参与因子Ⅸa对因子X的激活,使因子X被因子Ⅸa激活的速度大大提高。缺乏因子Ⅷ或因子Ⅸ时,凝血活酶生成减少,纤维蛋白凝块形成延迟,凝血时间延长,引起出血症状。vWF作为因子Ⅷ的载体对其起稳定作用,并参与血小板的黏附、聚集。vWF水平或功能降低时,可引起因子Ⅷ缺乏及出血倾向。
Ⅷ∶C 80%由肝窦内皮细胞合成,其余由脾、肺、肾、单核巨噬细胞等合成;其活性极不稳定,在4℃贮存24h后可丧失20%,Ⅷ∶C血浆含量5μg/L,活性50%~150%,半衰期8~12h。因子Ⅸ由肝脏合成,属于依赖维生素K的凝血因子,半衰期18~24h,血浆活性80%~120%。因子Ⅺ由肝脏合成,半衰期为40~48h,4℃下稳定,故本病患儿替代治疗时输入库存血浆即可补充因子Ⅺ。
临床表现
临床表现:
1.临床表现
(1)出血症状:为本病的主要表现,终身轻微损伤或手术后有持久出血倾向。
血友病甲、乙临床表现相似,出血症状出现越早病情越重。
血友病甲多在婴儿开始学爬、学走时发病,生后9个月内发病者少,偶见新生儿断脐时出血不止,轻症患儿可至成年后才发现。
血友病乙重型患儿少见,轻症患儿多,多在2岁内发病,少数迟至5~6岁。
(2)关节出血:是
血友病甲患儿的特殊表现之一,约见于75%的
血友病甲患者。常发生在运动及创伤后,婴儿多为踝关节受累,儿童以膝关节受累常见。出血前有轻度不适,继而关节局部红、肿、热、痛,活动受限。如出血量少,治疗及时,关节血肿可被吸收。但关节的反复出血常导致关节软骨破坏,关节腔变窄,关节周围肌肉萎缩形成慢性血友病性关节炎,甚至关节畸形、功能丧失。
(3)血友病肌肉出血和血肿:以下肢、前臂、臀部多见。深部血肿有相应部位疼痛、压迫症状。如出血量多,可引起休克、贫血、
黄疸及全身发热。皮下、齿龈、口腔及鼻黏膜易于受伤故为出血多发部位,但皮肤黏膜出血并非为本病的特征,皮肤瘀点、瘀斑少见。如出血发生在咽、喉易引起窒息。消化道出血、血尿亦常见,小儿血尿易误诊为“肾炎”。儿童脱牙或外科手术如拔牙、扁桃体摘除术等若不采取相应措施,会引起持久的渗血或出血。颅内出血少见,可以是
自发性,但通常由外伤引起,常危及生命。对伴有剧烈
头痛的血友病患儿应警惕颅内出血或硬膜下出血的可能。
血友病丙纯合子患儿有出血倾向,出血较轻,多发生在手术后或外伤后,
自发性出血少见;偶有皮肤黏膜出血,青春期女性可有
月经过多,出血程度与因子Ⅺ浓度无明显关系,患儿常合并因子V、因子Ⅶ等凝血因子缺乏。杂合子患儿无出血症状。
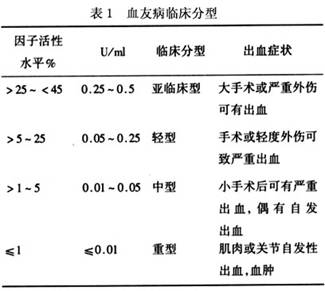
2.血友病临床分型
(1)重型:因子Ⅷ或因子Ⅸ活性<1%,多在1岁前出现
自发性出血,出血部位多且严重,反复关节内或深部组织(肌肉、内脏)出血,关节畸形多见。
(2)中间型:因子Ⅷ或因子Ⅸ活性为1%~5%,多在1~2岁时发病,创伤后可引起大出血,关节、肌肉出血多见,但反复发作次数少,很少在未成年前出现关节畸形。
自发性出血少见。
(3)轻型:因子Ⅷ或因子Ⅸ活性为6%~25%,多在2岁后发病,轻微损伤或手术后有出血不止,无
自发性出血及关节出血。
(4)亚临床型:因子Ⅷ或因子Ⅸ活性为26%~45%,仅在严重创伤、大手术后出血不止才发现本病,容易漏诊。
鉴别诊断
鉴别诊断:本病需注意与vWD相鉴别,后者因vWF质或量的异常引起血小板功能障碍,可借助于阿司匹林耐量试验、血小板对瑞斯托霉素的诱导无凝集反应及vWF因子抗原(vWF∶Ag)测定等鉴别。
1ml正常血浆所含的凝血因子的总量被定义为1个单位的因子。用活性的百分数表示因子的水平,即100%的水平(1U/ml)等于1ml正常血浆中因子的活性。根据FⅧ或FⅨ的水平将血友病分为4型(表1)。
1.
血友病甲和
血友病乙的鉴别诊断
血友病甲与
血友病乙的临床出血表现和家族遗传形式类似,两者APTT皆延长,难以鉴别。T
GT和纠正试验可以鉴别两者。最可靠的诊断手段是FⅧ∶C测定辅以vWF∶Ag测定和FⅨ∶C测定辅以FⅨ∶Ag测定。
2.与获得性因子缺乏症的鉴别 获得性因子缺乏症常见的是获得性FⅧ缺乏症,而获得性FⅨ缺乏症少见,临床表现和血友病相似,但出血程度较重,较多发生在妊娠女性、恶性肿瘤和免疫功能异常的患者。实验室检查APTT延长,小量的正常血浆不能纠正,测定抑制物滴度可以明确诊断。但应注意的是重型血友病患者长期应用因子替代治疗,可产生因子抑制物,
血友病甲发生因子抑制物的发生率远高于
血友病乙的发生率,此类患者治疗非常困难,出血死亡率高。
3.与血管性血友病的鉴别 此类疾病特别应与
血友病甲相鉴别,其特点是有或无家族史,有家族史者符合常染色体显性遗传或隐性遗传规律,男女均可发病,出血症状类似血友病。实验室检查:出血时间延长、阿司匹林耐量试验阳性、瑞斯托霉素(Ristocetin)诱导血小板聚集、APTT延长、FⅧ∶Ag下降或正常、vWF:Ag减低或正常(如正常需进一步检查是否为变异型)。
治疗
治疗:本病为先天性遗传性疾病,尚无根治疗法。
1.一般治疗 患儿自幼需加强护理,避免外伤及肌内注射,避免使用阿司匹林、非类固醇类抗感染药物及其他影响血小板聚集的药物。外科手术前、术中、术后应补充所缺乏的凝血因子。
2.局部治疗 皮肤外伤、鼻、齿龈出血可局部压迫止血,或用纤维蛋白泡沫、
吸收性明胶海绵等沾鲜血或新鲜
血浆敷于伤口处,大而深的伤口清创消毒后,以消毒棉球蘸
凝血酶、组织凝血活酶或新鲜
血浆涂于伤口,并加压包扎,局部冷敷。早期关节出血者,宜卧床休息,患肢夹板固定,置于功能位,冰袋和弹力绷带包扎。严重关节出血在补足所缺乏的因子和严密消毒后,可抽出积血,加压包扎。出血停止、肿痛消失后需进行适当体疗或牵引,防止关节畸形。
3.替代治疗 是治疗血友病的有效方法,目的是将患儿缺乏的因子提高到止血水平。
(1)输血及
血浆:血友病甲患者宜输新鲜血或新鲜
血浆,10ml/kg鲜血可提高患者
血浆中因子Ⅷ水平10%,疗效仅维持2天左右,适用于轻型和亚临床型患者。新鲜
血浆按1ml/kg输入可提高因子Ⅷ水平2%,每12小时1次。因子Ⅸ体外保存较稳定,血友病乙患者可以输库存5天以内的
血浆,每次输入量不宜过多,以10ml/kg为宜,每24小时1次。
血友病丙大手术或严重外伤时需用替代疗法,以鲜血或
血浆效果为佳。因子Ⅺ在体外较稳定,可用库存血。输入
血浆7~20ml/kg可使因子Ⅺ水平提高到25%~50%,手术前输
血浆30ml/kg,以后每天5ml/kg或隔天10ml/kg,维持至伤口愈合。
(2)冷沉淀物:系从冰冻新鲜
血浆中分出,包括原有
血浆中3%的
血浆蛋白、20%~85%的Ⅷ∶C和大量纤维蛋白原。各药厂产品及剂量不一,用前应详细参阅说明书。通常以400ml血中冷沉淀物含因子Ⅷ100单位(U)计算(1U=1ml正常新鲜
血浆所含因子Ⅷ的量),输入1U/kg可提高
血浆中因子Ⅷ水平2%。用量因出血轻重、部位不同而有差异,适用于轻、中型血友病甲(表2)。
(3)因子Ⅷ浓缩剂:多用人
血浆冻干浓缩剂,所需因子Ⅷ的剂量按以下公式计算;所需剂量(U)=体重(kg)×所需提高的水平(%)×0.5,每12小时1次。具体用法见表1。
(4)因子Ⅸ浓缩剂:可按1U/kg输入,每24小时1次。
(5)重组
抗血友病因子:基因工程制备的重组因子Ⅷ,效果好,反应少,不传播病毒性疾病,适用于血友病甲。输入1U/kg可提高因子Ⅷ 2.7%。
替代治疗不良反应是约3.6%~25%血友病甲患儿产生因子Ⅷ抗体;1%血友病乙患儿产生因子Ⅸ抗体。经常使用血液制品,使患儿易并发肝炎、艾滋病。足量因子Ⅷ制品治疗后,仍不能控制出血或反而加重,提示有因子Ⅷ抗体存在,机体对外源性因子Ⅷ产生免疫反应。根据患者免疫应答反应的不同分为高反应者(血中抗体效价高)和低反应者(抗体效价低)。对这些患者治疗的目的是制止出血、去除抗体。对低反应者可大剂量输入因子Ⅷ,每次50~100U/kg,8~12h 1次,部分用于中和抗体,部分用于维持止血水平。高反应者的治疗包括:①持续性输入因子Ⅷ,每次100U/ml,2次/d,疗程7~10天。②输入
凝血酶原复合物(75U/kg,2次/d)或活化
凝血酶原复合物(75U/kg,每6~12小时1次),可改善止血功能且止血效果与因子Ⅷ抗体效价无关,但有诱发高凝和血栓的危险。③猪因子Ⅷ浓缩剂,每天20~100U/kg静滴,疗程2~4周。④重组因子Ⅶa,可与组织因子共同作用激活X因子,促进凝血活酶的形成。按70~100μg/kg,每2~4小时静脉给药1次。⑤免疫抑制药,如环磷酰胺。⑥
血浆置换术,作为辅助治疗措施,清除因子Ⅷ抗体。
4.药物治疗
(1)
凝血酶原复合物:含有因子Ⅸ,适用于血友病乙中、重度出血患者。剂量同因子Ⅷ浓缩剂,每24小时1次,直至达到止血效果。新生儿慎用,因可诱发血栓性栓塞。
(2)1-脱氨-8-
精氨酸加压素(DDAVP):可提高因子Ⅷ水平4倍,是轻型血友病患者有效的替代治疗。剂量为0.3~0.4μg/kg,溶于20ml生理盐水中缓慢静注,2次/d,每疗程2~5次;或用滴鼻剂,体重低于50kg者,150μg/次滴鼻;超过50kg者,300μg/次滴鼻,2次/d。不良反应有轻微心率加快,颜面潮红。应避免摄入过多液体并监测尿液中药物浓度,以防止低钠血症和脑水肿。
(3)抗纤溶疗法:保护少量已形成的凝血块不被溶解,用于黏膜出血及拔牙术后的替代治疗。
氨基己酸(
EACA)0.1g/kg,4次/d口服。
氨甲环酸(AMCA)5mg/kg,3次/d口服;静脉注射5mg/kg,1~2次/d。
上述两种药物应在DDAVP治疗或因子Ⅶ替代治疗的情况下使用,连用7天或直至血止。忌与
凝血酶原复合物同用,血尿患者不宜使用。
(4)其他:达那唑(danazol)是一种合成的17-烷基化雄性激素,男性化作用弱,可提高因子Ⅶ浓度,降低出血倾向,疗效逊于替代疗法。雷尼替丁(Rinitidine)0.1~0.15g/d口服,剂量随年龄递增,疗程4天以上,可提高因子Ⅷ∶C的活性,适用于轻、中型血友病患者,重型患者常无效。
5.血友病乙基因治疗 我国于1991年以反转录病毒为载体,进行世界首次血友病乙基因治疗的临床工期实验,取得了成功。
6.家庭治疗 欧洲20个血友病治疗中心调查发现52%的患儿是重型血友病,只有29%的是轻型血友病,而在血友病总体人群调查显示轻型血友病为50%~55%,多数学者认为在很多国家中很多轻型血友病患儿容易被漏诊,因此很多轻型血友病患儿得不到及时治疗,在发展中国家此情况尤为突出。对于血友病患儿的治疗,基因治疗可能是治愈该病的惟一方法,但是基因治疗目前仍不能广泛地应用于临床。目前预防性治疗被认为是血友病治疗的金标准,然而由于其耗费昂贵,此项治疗在发展中国家尚难开展。
家庭治疗在血友病治疗史中具有划时代的意义,目前在国外已广泛推广。除有抑制性抗体、病情不稳定、小于3岁的患儿外,均可使用家庭治疗。血友病患者及其家属应接受有关疾病的病理、生理、诊断以及治疗知识的教育,并在专业医师的指导下进行注射技术的培训,掌握熟练的操作技术,以便在患者出血时能够尽早实施因子治疗,以防止大血肿的形成、畸形或残疾的发生。并应该有专业医师定期随访、咨询和指导。近几年来国外已经开展家庭预防性替代治疗,并取得了良好的效果。
7.围术期的治疗 血友病患者凡行外科手术,不论是择期手术还是急诊手术,都应做好充分的术前准备。术前必须明确诊断,检测是否存在因子抑制物,并准备充足的血源和因子制剂。在术中和术后要有适当的监测和康复措施。
血友病患者手术前应给予足量的替代因子(FⅧ或FⅨ)。对于大手术,术前1h应确保因子水平在50%~80%,然后因子水平维持在30%~50%,水平应保持10~14天。口腔手术前同样要求因子水平在50%~80%。为防止发生出血,术后可联合抗纤溶药物治疗7~10天。若术后伤口发生感染,或手术范围广泛,损伤较大,则应延长替代治疗时间。轻型血友病A患者,术前可使用DDAVP,最好与FⅧ联合使用;而轻型的血友病B患者,只能用FⅨ替代治疗。
预防
预防:加强血友病携带者的检测,对血友病家族中的孕妇进行产前诊断,血友病胎儿应终止妊娠,无疑会降低血友病的发病率。
根据本组疾病的遗传方式,对患者的家族成员需进行筛查,以确定其中病人和携带者,并对他们进行有关本组疾病的遗传咨询,使他们了解遗传规律。对家族中的孕妇要采用基因分析法进行产前诊断,如确定胎儿为
血友病甲患者,可及时终止妊娠。
预防出血应自幼养成安静生活习惯,以减少和避免外伤出血,尽可能避免肌内注射,如因患外科疾病需做手术治疗,应注意在术前、术中和术后输血或补充所缺乏的凝血因子。